Keywords
Hemophagocytic syndrome; Lymphohistiocytosis; Hemophagocytic; Pancytopenia; Infant
Introducción
El Síndrome hemofagocítico (SH) llamado también Linfohistiocitosis hemofagocítica (LHH) o Síndrome de activación macrofágica (SAM), es un trastorno poco frecuente, que se define como una activación inmuno-patológica que lleva a la alteración reactiva del sistema inmune-mononuclear, provocando sobre estimulación, proliferación y migración ectópica de linfocitos T, que lleva a la producción excesiva de citocinas (interferón-gamma, factor de necrosis tumoral alfa, interleucinas (IL-1, IL-6, IL-10, IL-12, IL-18) y factor estimulante de colonias de macrófagos), resultando en la activación de histiocitos con la subsecuente hemofagocitosis [1,2]. La incidencia se estima en 1.2 casos por millón de individuos al año, aunque probablemente es una cifra subestimada, ya que en muchas ocasiones es un diagnóstico que no se sospecha1. Se ha identificado que no tiene predilección por edad, raza o género, pero sigue un patrón estacional, ocurre más frecuentemente en verano [3].
La LHH se caracteriza por signos y síntomas clínicos de un proceso inflamatorio excesivo. Se reconoció por primera vez como un trastorno inmunológico desregulatorio familiar de la infancia a la llamada “Reticulosis Hemofagocítica Familiar” esto en 1,952. Más tarde, fue descrita la LHH como un trastorno familiar y esporádico, este último asociado con infecciones, neoplasias malignas o trastornos reumatológicos. La base inmunológica se sospechó debido a su naturaleza inflamatoria y el hallazgo de deficiencias de citotoxinas y otras anormalidades inmunes en pacientes con LHH [4]. La primera visión genética en la etiología de LHH fue en 1,999 con el descubrimiento de mutaciones de las perforinas en algunos pacientes afectados [5].
Se ha descrito una forma primaria o genética (familiar) y una forma secundaria o adquirida (reactiva). La forma familiar es de herencia autosómica recesiva, por lo que la historia familiar a menudo es negativa. La forma secundaria es el resultado de una reacción inmunológica intensa e incontrolable causada por procesos infecciosos, neoplásicos y autoinmunitarios (Tabla 1) [1,6,7].
Primaria
Linfohistiocitosis hemofagocítica familiar (con sus 4 subtipos: LHHF 1-4)
Secundarias
Infecciones
Víricas: Citomegalovirus, virus herpes, adenovirus, parvovirus, coxsackie, virus Epstein-Barr, VIH, varicela zóster, hepatitis A,B,C.
Bacterianas:Salmonella, enterobacterias, Rickettsia coronii, Mycobacterium tuberculosis, Mycoplasma pneumoniae, Coxiella burnetti, espiroquetas (leptospira spp, Treponema pallidum).
Parasitarias: Leishmania spp, Plasmodium falciparum, Toxoplasma gondii, Babesia microti.
Fúngicas:Aspergillus, Histoplasma capsulatum, Candida albicans, Cryptococcus neoformans.
Enfermedades Autoinmunitarias
Artritis reumática juvenil idiopática, Lupus eritematoso sistémico, paniculitis histiomacroesofágica.
Enfermedades Malignas
Leucemias o Linfomas T o NK, tumores germinales mediastinales.
Fármacos
Emulsiones lipídicas de nutrición parenteral, sales de oro, salazopirina, ácido acetilsalicílico, indometacina. |
LHHF: Linfohistiocitosis Hemofagocítica Familiar.
Tabla 1: Clasificación: Formas de síndrome hemofagocítico (Kumakura 2005).
Presentación de caso
Se presenta el caso de lactante mayor femenina de 2 años de edad, procedente de zona rural del departamento de Lempira, Honduras, con antecedente de fiebre de 5 días de evolución no cuantificada, subjetivamente elevada, intermitente, de predominio nocturno, con escalofríos. Acompañado de rash de 3 días de evolución y tinte ictérico que inició en escleras y mucosa oral, y que posteriormente se generalizó. Afirma coluria, astenia, adinamia e hiporexia de 2 semanas de evolución. Al examen físico, presentó signos vitales de P/A:96/55 mmHg, PAM:70, FC:135x`, pulso:135x`, FR:22x`, T°:39°C. Cardiovascular: soplo sistólico grado II/VI, abdomen: hepatomegalia ± 5 cm por debajo del reborde costal derecho, y esplenomegalia ± 3 cm por debajo del reborde costal izquierdo dolorosa a la palpación profunda. Piel con tinte ictérico generalizado (+++). Resto del examen físico sin alteraciones.
Se realizó hemograma que reportó pancitopenia (leucopenia, trombocitopenia y anemia). La química sanguínea reportó: hipertransaminasemia, hiperbilirrubinemia a expensas de bilirrubina directa, fosfatasa alcalina aumentada, hiperuricemia, hiperferritinemia, hipoproteinemia, hiperlipidemia mixta, prolongación de los tiempos de coagulación, trastornos hidroelectrolíticos (hiponatremia, hipokalemia, hipocalcemia), reactantes de fase aguda positivos (PCR, VES), hipoinmunoglobulinemia (IgG), urocultivo que reportó la presencia de E. Coli>105 Unidades formadoras de colonia/ ml, y aspirado de medula ósea que se evidenció datos de hemofagocitosis (Tabla 2).
| Hallazgo Laboratorial |
Resultado |
Valor normal |
| Glóbulos rojos (x 106/ul) |
4.1 |
(4.2–5.4) |
| Hemoglobina (g/dl) |
7.3 |
(12-16) |
| Hematocrito (%) |
21.5 |
(37-47) |
| Plaquetas (x 103/ul) |
1,35,000 |
(150-500) |
| Glóbulos blancos (x 103/ul) |
1,290 |
(5.2-12.4) |
| Neutrófilos (x 103/ul) |
0.27 |
(1.9-8) |
| Linfocitos (x 103/ul) |
0.72 |
(0.9-5.2) |
| TSGO (U/L) |
315 |
15-37 |
| TSGP (U/L) |
170 |
12-78 |
| Fosfatasa alcalina (U/L) |
1154 |
48-116 |
| Bilirrubina Total (mg/dl) |
9.7 |
0.2-1.0 |
| Bilirrubina directa (mg/dl) |
8.4 |
0.0-0.2 |
| Bilirrubina indirecta (mg/dl) |
1.3 |
0.0-0.2 |
| Ácido úrico (mg/dl) |
1.3 |
3.5-7.2 |
| Ferritina (ng/dl) |
>1500 |
28-397 |
| Colesterol total (mg/dl) |
247 |
0-200 |
| Triglicéridos (mg/dl) |
522 |
30-150 |
| Tiempo protrombina (seg) |
20 |
10-15 |
| Tiempo tromboplastina parcial (seg) |
80 |
30-35 |
| Sodio (mmol/L) |
129 |
135-145 |
| Potasio (mmol/L) |
4.1 |
3.4-5.1 |
| Calcio (mmol/L) |
6.9 |
8.5-10.1 |
| PCR (mg/dl) |
48 |
0.1-1 |
| VES (ml/hr) |
10 |
0-10 |
| Inmunoglobulina G (mg/dl) |
41 |
47-180 |
TSGO: Aspartato-aminotransferasa; TSGP: Alanino-aminotransferasa; PCR: Proteína C reactiva; VES: Velocidad de eritrosedimentación
Tabla 2: Hallazgos de laboratorio.
A su vez se realizó ultrasonido abdominal total que evidenció: hepato-esplenomegalia y vesícula contraída sin presencia de litos en su interior. Resto de ultrasonido dentro de parámetros normales (Figura 1).
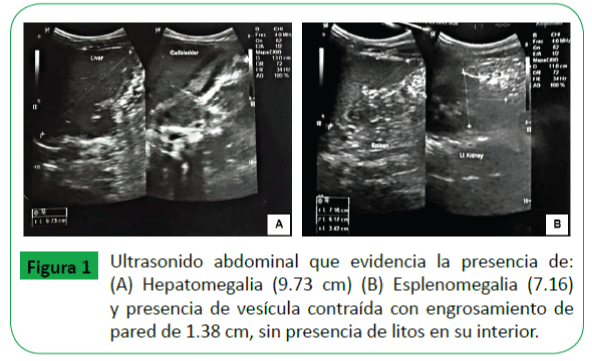
Figura 1: Ultrasonido abdominal que evidencia la presencia de: (A) Hepatomegalia (9.73 cm) (B) Esplenomegalia (7.16) y presencia de vesícula contraída con engrosamiento de pared de 1.38 cm, sin presencia de litos en su interior.
El tratamiento inicial consistió en cobertura antibiótica con cefalosporinas de tercera generación (ceftriaxona) y betalactámicos (oxacilina) por un periodo de 10 días. Pese a los resultados del urocultivo positivo y antibiograma específico, la paciente persistió con episodios febriles, por lo que se realiza cambio de cobertura antibiótica a carbapenémicos (imipenem). Asimismo, se inició tratamiento inmunomodulador con corticoides (pulsos de metilprednisolona) y se administró gammaglobulina intravenosa (por 48 hrs). Se realizaron transfusiones con glóbulos rojos empacados para corregir anemia microcítica hipocrómica y se aplicó vitamina k por 3 días para corregir el defecto de los tiempos de coagulación. La paciente curso con mejoría muy significativa, continuando ingresada hasta culminar su cobertura antibiótica y posteriormente egresar a su domicilio.
Discusión
Las principales manifestaciones clínicas que acompañan al SH son: fiebre prolongada que no responde a antibióticos, citopenias, esplenomegalia, hepatomegalia, hemofagocitosis, hipertrigliceridemia y/o hipofibrinogenemia [1], puede acompañarse también con coagulopatía e hiperferritinemia [1,3,8-10]. Otras manifestaciones clínicas menos frecuentes son las adenopatías, el rash, los edemas y la ictericia6. Signos y síntomas que presentó nuestra paciente, además de la presencia de síntomas constitucionales (astenia, adinamia, hiporexia). Los hallazgos de laboratorio incluyen citopenias, generalmente comienza con trombocitopenia que evoluciona a pancitopenia, hiperferritinemia, elevación de transaminasas, hipofibrinogenemia, hipertrigliceridemia, hipoalbuminemia e hiponatremia. Datos que en este caso fueron confirmados mediante laboratotorio [1,6,9].
Dentro de los hallazgos histológicos se encuentran células hemofagocíticas en la biopsia de medula ósea, bazo y ganglios linfáticos [11]. En este caso, se confirmó la presencia de hemofagocitosis mediante frotis de sangre periférica y aspirado de medula ósea.
El diagnóstico del SH se establece por la presencia de cinco de los ocho criterios diagnósticos aceptados actualmente, propuestos por la Histiocyte Society (Tabla 3) [1,12,13]. La sospecha precoz es fundamental, en especial si existe causa potencial. La concurrencia de ≥ 5 criterios, puede no ocurrir inmediatamente e ir apareciendo de forma paulatina [7,14]. En este caso la paciente presentó 6 criterios diagnósticos, los cuales se confirmaron mediante la evaluación clínica, exámenes laboratoriales y de gabinete.
Enfermedad familiar o defecto genético conocido de SH familiar, o presentar 5 de los 8 siguientes:
- Fiebre ≥ 38,5°C
- Esplenomegalia
- Citopenia que afecte a 2 o más series (Hb<9 g/dl [niños<4 semanas, Hb<10 g/dl], plaquetas<100.000/L, neutrófilos<1.000/L)
- Hipertrigliceridemia (>265 mg/dl) y/o Hipofibrinogenemia (<150 mg/dl)
- Hemofagocitosis en médula ósea, bazo, hígado o ganglios, sin evidencia de malignidad.
- Actividad citotóxica de células NK baja o ausente
- Hiperferritinemia (> 500 ng/ml)
- sCD25 soluble elevado (sIL-2R>2.400 U/ml) |
Fuente: HLH-2004, en Janka
Tabla 3: Criterios diagnósticos del síndrome hemofagocítico.
En la mayoría de los casos se reporta una activación anómala y persistente de los macrófagos, así como una alteración importante de la inmunidad por parte de los linfocitos Natural Killers (NK) y T, en especial, una inadecuada respuesta de los linfocitos Th1 con una mayor producción de citocinas pro inflamatorias. Esto conduce a un daño tisular de rápida progresión y finalmente al fracaso multiorgánico [7]. Esto se presenta con mayor frecuencia en la forma secundaria o reactiva, como es ejemplo este caso, debido a una respuesta inmune exagerada frente a un antígeno.
Dentro de las causas secundarias de SH reactivo se encuentran las infecciones bacterianas, entre las cuales se mencionan: Salmonella, enterobacterias, Rickettsia Coronii, Mycobacterium tuberculosis, Mycoplasma pneumoniae, Coxiella burnetti, espiroquetas (Leptospira sp, Treponema pallidum) [7]. Un estudio de 16 casos clínicos realizado en el Hospital de Cabueñes (Gijón) evidenció que la etiología más habitual fue la infecciosa (viral: VIH, bacteriana: Mycobacterium avium intracellulare, staphylococcus aureus, legionella y mycoplasma). A su vez se demostró la presencia de coinfección confirmada por urocultivo evidenciando a klebsiella como agente causal [15]. Pese a que se realizaron múltiples serologías en busca de antígenos virales, las cuales se reportaron negativas, se atribuyó a que la causa desencadenante se debía una infección del tracto urinario por Escherichia coli, resultado que se confirmó por urocultivo. Debido a que el examen general de orina no reportó anormalidades en dos ocasiones.
El diagnóstico diferencial es fundamental para el tratamiento, pronostico y consejo genético [16]. El SH se puede presentar como fiebre de origen desconocido, hepatitis, falla hepática aguda, enfermedad de Kawasaki y anormalidades neurológicas. Tiene una alta mortalidad, con una supervivencia de 2 meses sin tratamiento, por lo que es prioritario iniciar manejo en cuanto se establezca el diagnóstico. 1 Se han descrito entre los factores de mal pronóstico: edad>30 años, hiperferritinemia>10,000 ng/ dl, trombocitopenia y presencia de Coagulación intravascular diseminada (CID) [4]. En este caso encontramos la presencia de hiperferritinemia>1500 ng/dl y trombocitopenia.
El tratamiento se basa en suprimir la activación exagerada del sistema inmune. En los casos de SHF familiar se basa en el protocolo HLH 043, un protocolo pediátrico, que se usa como puente para trasplante alogénico. En el sistema público de Honduras, esta alternativa terapéutica parece poco factible en vista de ser un país en vías de desarrollo [17]. Se han utilizado diferentes tratamientos inmunomoduladores en el SH secundario: corticoides (en especial dexametasona por su capacidad para inhibir la expresión de citocinas y por su habilidad de cruzar la barrera hematoencefálica, produciendo diferenciación de células dendríticas), inmunoglobulinas, globulina antitimocítica, ciclosporina A, etopósido (por su actividad frente a las células infectadas presentadoras de antígenos; con mejoría de la supervivencia) y alemtuzumab. Los primeros días/semanas del tratamiento pueden coincidir paradójicamente con los de mayor sintomatología, incluyendo el fracaso multiorgánico y la necesidad de soporte vital avanzado en unidades de críticos [1,4,17,18].
Conflicto de Interés
Los autores declaran no tener conflictos de interés en la publicación del presente artículo.
11267
References
- Bautista EKA, FossasGP, Rodríguez LE (2013)Síndromehemofagocítico.Conceptosactuales.GacetaMédica de México 149:431-437.
- DubucECA, EcenarroUM, VillalbaMC, CáceresAV, Rubio HN, et al. (2014)Síndromehemofagocíticocomomanifestaciónclínicainicial del lupus eritematososistémico. ReumatolClin 10: 321-324.
- Carrillo-Esper R, Rivero-Martínez JA, Zepeda-Mendoza AD (2014)Síndromehemofagocíticoasociado con virus de la influenza A H1N1. Med IntMéx30: 738-744.
- Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL (2011) How I treat hemophagocyticlymphohistiocytosis.Blood 118: 4041-4052.
- Stepp SE, Dufourcq-Lagelouse R, Le Deist F,Bhawan S, Certain S, et al. (1999)Perforin gene defects in familial hemophagocyticlymphohistiocytosis. Science 286:1957-1959.
- Pérez-Martínez A (2013)Síndromeshemofagocíticos (I): concepto, clasificación, fisiopatología y clínica. AnPediatrContin. 11:237-244.
- González FO, FernándezIM, Gómez RA, Redondo UC, GarcíaCML (2014)Síndromehemofagocíticoasociado a virus de Epstein-Barr. Descripción de uncaso y tratamiento en la Unidad de Reanimación. Rev EspAnestesiolReanim61:571-574.
- BacarrezaNJJ, LópezML, AlcorezaNPJR (2011)Síndromehemofagocíticoasociadoainfección viral porcitomegalovirus. Med Intensiva 35: 189-192.
- Berdugo LP, Rodríguez ZN, Tordecilla CJ, Soto AV (2005)Síndromehemofagocíticosecundario en pediatría. Experienciaclínica en ochocasos. Rev ChilPediatr 76: 397-403.
- UrgellésSA (2002)Síndromeshemofagocíticos: pensar en ellos... porqueexisten. An EspPediatr 56: 95-98.
- DubucEC, CáceresAV, EcenarroUM, Aguirre EN, Rubio HI, et al. (2015)Síndrome de activaciónmacrofágicasecundario a enfermedadesautoinmunes, hematológicas, infecciosas y oncológicas. Serie de 13 casosclínicos y unarevisiónbibliográfica.ReumatolClin11:139-143.
- Henter JI, Elinder G, Ost A (1991) Diagnostic guidelines for hemophagocyticlymphohistiocytosis. The FHL Study Group of the Histiocyte Society.Semin Oncology18:29-33.
- Henter JI, Horne A, Arico M,Egeler RM, Filipovich AH, et al. (2007) HLH-2004: diagnostic and therapeu- tic guidelines for hemophagocyticlymphohistiocytosis. Pediatr Blood Cancer48:124-131.
- GarcíaHR, VillenaBP, GuillénaMS, BernabéuPA. Síndromehemofagocítico en paciente con polimialgiareumática. ReumatolClin12:50-51.
- Barbón SR, González-García ME (2009)Síndromehemofagocítico. Estudio de 16 casos. Med Clin (Barc)133:74-75.
- Martínez I, Fernández L, Valentín J, Castillo C, Chamorro C, et al. (2015) La actividadcitotóxica de lascélulas natural killer comoherramientadiagnóstica en pacientespediátricoscríticos con sospecha de síndromehemofagocítico. Med Intensiva39:213-221.
- Peña C, Valladares X, Cabrera ME (2013)Síndromehemofagocíticosecundario: reporte de 5 casos. Rev Med Chile 141: 1475-1479.
- Madkaikar M, Shabrish S, Desai M (2016) Current Updates on Classification, Diagnosis and Treatment of HemophagocyticLymphohistiocytosis (HLH). Indian J Pediatr 1: 1-10.

