Babak Abdolkarimi1, Soheila Zareifar2 and Mansureh Shokripoor3
1Department of Pediatric Oncology/Hematology, Lorestan University of Medical Sciences, Khoramabad, Iran
2Hematology/Oncology Department, Amir Oncology Hospital, Shiraz University of Medical Sciences, Shiraz, Iran
3Pathology Department, Shiraz University of Medical Sciences, Shiraz, Iran
Corresponding Author:
Babak Abdolkarimi
Assistant Professor
Department of Pediatric Oncology/Hematology
Lorestan University of Medical Sciences, Khoramabad, Iran
Tel: +989183605274
E-mail: b.abdolkarimi@yahoo.com
Received date: July 16, 2016; Accepted date: July 29, 2016; Published date: August 02, 2016
Citation: Abdolkarimi B, Zareifar S, Shokripoor M. Successful Treatment of Primary Intracranial Malignant Peripheral Nerve Sheath Tumor in Iranian Child: Case Report. J Neurol Neurosci. 2016, 7:4.
Keywords
Malignant peripheral nerve sheath tumor (MPNST); Neurofibroma; Malignant schwannoma
Introduction
A malignant peripheral nerve sheath tumor (MPNST) or malignant schwannoma, neurofibrosarcoma, and neurosarcoma arises is a form of non-rhabdoid sarcoma arise from connective tissue surrounding nerves [1].
About half the cases are diagnosed in people with neurofibromatosis and lifetime risk for an MPNST in patients with neurofibromatosis type 1 is 8-13% [2]. MPNST can occur anywhere in the body, but most often occur in the deep tissue of the arms, legs and trunk [3]. These tumors occur more frequently in neurofibromatosis and persons undergone radiation therapy for cancer. However, most people have no risk factors for the disease [4].
Intracranial MPNST is a rare and highly malignant tumor. The incidence of this subtype is 0.001%. The prognosis of the tumor is extremely poor and can be graded to IV of WHO classification [5]. MPNST is typically treated with but, surgery for incomplete unresected tumors after surgery chemotherapy or radiation therapy should be considered [6].
Case Presentation
A 3.5-year-old female presented to the pediatric out-patient department with complaints of headache and swelling of left side of skull, which was progressive over a period of 3-months.
On examination the patient was conscious and coherent. She had not cranial nerve palsy but, muscle force in the right- sided limb was 3/5. Right plantar showed an extensor response. There wasn’t any constitutional symptom such as fever detected in patient.
Trigeminal Nerve involvement symptoms include transient hearing loss (objective assessment), residual facial pain, and paresthesia of the left side of the face were not detected.
CBC count, bilateral bone marrow aspiration biopsy, Liver function tests, including measurement of lactic acid dehydrogenase (LDH), aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase, and bilirubin levels and Renal function tests, including measurements of BUN and creatinine levels and Blood electrolyte and chemistry, including evaluation of sodium, potassium, chlorine, carbon dioxide, calcium, phosphorous, and albumin values were normal.
Metastatic disease of the liver didn’t detect in abdominopelvic CT scan. Thoracic CT scan also was normal.
Her MRI of head showed a large left temporal enhancing lesion with local bone involvement but spinal MRI was normal (Figure 1). Magnetic resonance imaging (MRI) of the orbits and brain with and without contrast enhancement in the axial plane showed a large soft-tissue mass at the lateral side of the optic nerve. The tumor invaded the lateral rectus muscle and right temporal pole Patient was underwent craniotomy and total excision of the tumor (Figure 2).
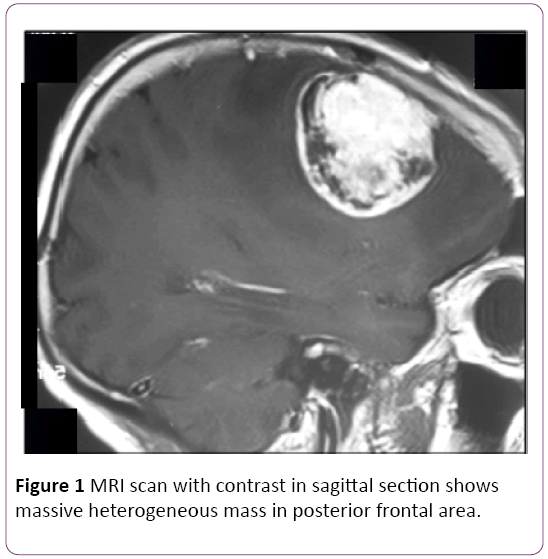
Figure 1: MRI scan with contrast in sagittal section shows massive heterogeneous mass in posterior frontal area.
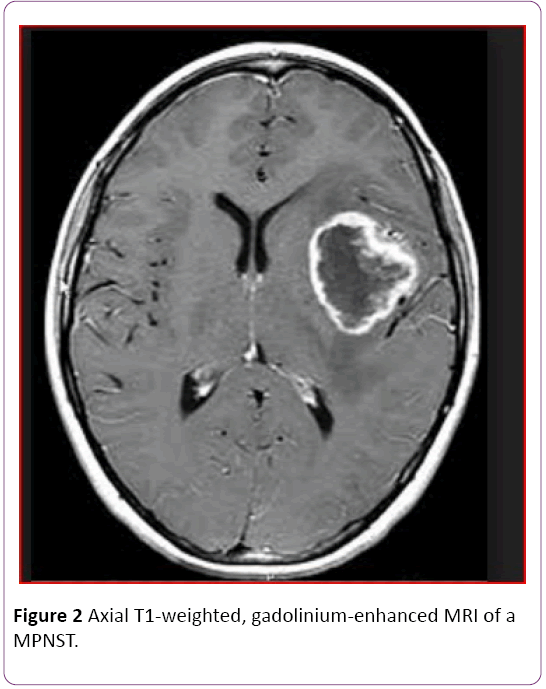
Figure 2: Axial T1-weighted, gadolinium-enhanced MRI of a MPNST.
The tumor was highly vascularized and had eroded the sphenoid bones, sphenoid wing, dura, and temporal pole. The lesion was removed totally with microsurgical technique. Duroplasty was performed. Microscopically, malignant differentiation and infiltration into the lateral rectus muscle were observed.
Histologically, tumor had foci of extremely high cellularity, pleomorphism and mitosis with fascicular pattern, hyperchromatic nuclei, and numerous mitotic and extensive necroses (Figure 3). Immunohistochemically, the tumor cells were positive for S-100 protein. Immunostains were negative for GFAP, EMA, CD117, CD34 and Myo-D1. The immunostain for S- 100 was focal positivity.
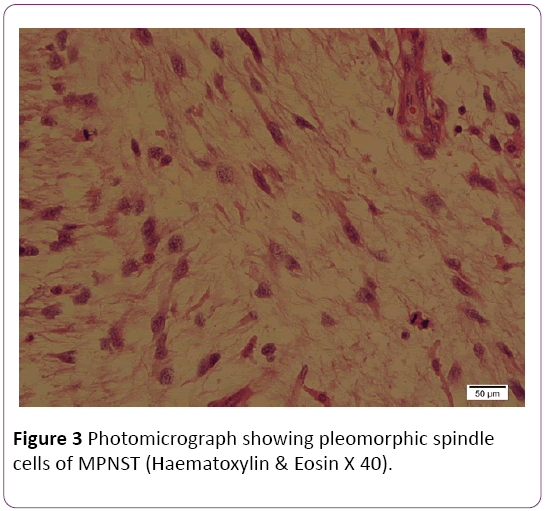
Figure 3: Photomicrograph showing pleomorphic spindle cells of MPNST (Haematoxylin & Eosin X 40).
She was treated by high risk Rhabdomyosarcoma chemotherapy protocol includes: Vincristine 1.5 mg/m2, weekly for first 12 weeks and then with each cycle. Actinomycin D 0.045 mg/kg IV push Actinomycin is omitted during radiation therapy. This regimen is typically given for 12– 14 cycles lasting 8–10 months. Ifosfamide 2 g/m2 + Mesna from day 1 to 3. This regimen was prescribed for 10 cycles every 21 days. Radiotherapy was delivered for patient with conventional fractionation (2.0 Gy daily for 5 days a week).
Discussion
Most MPNST arise from the nerve plexuses that distribute nerves into the peripheral area such as brachial and lumbar plexuses but our patient was a intracranial schwannoma [7]. Only 15 cases of intracranial MPNST’s are reported in the world literature until 2016 among children during our electronic search in PubMed. This tumor represents 8% of all primary brain tumors. It usually develops from the Schwann cells of a cranial nerve. Hemispheric intraparenchymal localisations are quite rare. Intracerebellar localization is even by more exceptional. In 1977, one case was reported by Komminoth. Since then only one other case has been published in 1987 by Sarkar [8].
Glioblastoma multiform and gliosarcoma, anaplastic meningioma needs to be ruled out but these tumors are rare in childhood [7].
Postoperative radiotherapy and chemotherapy may delay the onset of recurrence and is recommended for MPNSTs in NF-1 patients but in other patients role of adjuvant radiotherapy remains controversial even may be implicated in the pathogenesis of MPNSTs [9]. Adjuvant radiotherapy with stereotactic strategy provides the possibility to apply higher radiation doses even with nearby sensitive structures such as the brainstem. Karami et al. showed stabilization of this aggressive tumor with the combined use of conventional fractioned radiotherapy and stereotactic gamma knife radiosurgery [10].
MPNST’s traditionally chemotherapy includes doxorubicin and ifosfamide but it has partial response rates of 20-25% and patients with von Recklinghausen’s disease have 30% - 39% response with 5-year survival rates from 37.6% to 65.7%. Prognosis and overall survival for MPNST of the head and neck, a 5-year cause-specific mortality rate of 56% to 67% has been reported [9].
In our patient response with surgery and radiation therapy and chemotherapy was complete and our patient after 2 years end of treatment hasn’t relapse or neurologic dysfunction.
Also management of intracranial MPNSTs is firstly surgical with adjuvant controversial radiotherapy and chemotherapy [11].
Intracranial MPNSTs are a rare entity. Compared to MPNSTs from all body sites, it arise more frequently associated with radiation exposure but, our patient hadn’t any focus of tumor on evaluations and exposure to radiation before disease.
The association between intracranial MPNSTs and NF2 is as strong as its association with NF1, if not even stronger probably NF2 was is suspected but, in our patient didn’t exist any evidence of NF1 and NF2 [11]. Ionizing radiation is an established risk factor for MPNST development, especially in susceptible patients such as those with NF1.
While patients with NF1 represent a population with genetic susceptibility to radiation-induced tumors, the pathogenesis of intracerebral MPNSTs remains poorly understood [12].
There are different chemotherapeutic regimens as a neoadjuvant are used in this subgroup.
Various chemotherapy schedules used include VACA or VAC/ CAV regimens including vincristine, doxorubicin, cyclophosphamide, and dactinomycin. Ifosfamide replaced cyclophosphamide in VAIA and IVA regimens. Carboplatin and etoposide were used in the CEVAIE schedule or as two-drug therapy.
In addition, a combination of cisplatin (CDDP) and etoposide or other regimens (without ifosfamide or cyclophosphamide) were suggested [10].
Conclusion
Intracranial MPNST is the most malignant subtype of MPNST with a poor prognosis.
The mainstay of treatment is radical surgical resection with adjuvant radiotherapy or chemotherapy. For these tumors close follow-up is mandatory especially in patients with NF1. MPNSTS are currently treated as other soft-tissue sarcomas, because they are too rare to perform trials with a sufficient number of patients. Overall survival with MPNSTS is poor, and the usual chemotherapy used for soft-tissue sarcomas does not improve the outcome. Recent advances in the molecular biology of MPNSTS may provide new targeted therapies [13,14].
Conflict of Interest
The authors declare that they have no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
10580
References
- Evans DGR, Baser ME, McGaughran J, Sharif S, Howard E, et al. (2002) Malignant peripheral nerve sheath tumours in neurofibromatosis-1. Journal of Medical Genetics 39: 311-314.
- Walker L, Thompson D, Easton D, Ponder B, Ponder M, et al. (2006) A prospective study of neurofibromatosis type-1 cancer incidence in the UK. British Journal of Cancer 95: 233-238.
- Little MP, De Vathaire F, Shamsaldin A, Oberlin O, Campbell S, et al. (1998) Risks of brain tumour following treatment for cancer in childhood: modification by genetic factors, radiotherapy and chemotherapy. Int J Cancer 78: 269-275.
- Stark AM, Buhl R, Hugo HH, Mehdorn HM (2001) Malignant peripheral nerve sheath tumours-report of 8 cases and review of the literature. ActaNeurochir 143: 357-363.
- OzdalB, Murat OZ, KorkmazE, AtaogluO, Gungor T, et al. (2014) Malignant peripheral nerve sheath tumor of the vulva, an unusual differential diagnosis for vulvar mass. Int J Surg Case Rep 5: 793–795
- Byung SL, Young GK, Dong HK, Mou SL (2013) A long-term survival case of a primary malignant intracerebral nerve sheath tumor. J Korean NeurosurgSoc 54: 261–264.
- Benazza A, Houtteville JP, Chapon F, Khouri S, Hubert P (1989) Intracerebellarschwannoma. Apropos of a case. Review of the literature. Neurochirurgie 35: 246-251.
- Charles JF, Samir T, Aaron JK, Stephanie R, Jacqueline VA, et al. (2012) Perioperative management of neurofibromatosis type-1. Ochsner J 12: 111–121.
- Modesto C, Andrea F, Adrian M, Ilaria Z, Michela C, et al. (2005) Pediatric malignant peripheral nerve sheath tumor: The Italian and German Soft Tissue Sarcoma 23: 33.
- Saliba I (2013) Updates on the diagnosis and treatment of intracranial nerve malignant peripheral nerve sheath tumors. OncoTargets and therapy 6: 459-470.
- Carli M, Morgan M, Bisogno G (1995) Malignant peripheral nerve sheath tumors in childhood (MPNST): A combined experience of the Italian and German co-operative studies. Med PediatrOncol 25: 243.
- Gousias K, Bostrom J, Kovacs A, Niehusmann P, Wagner I, et al. (2010) Factors of influence upon overall survival in the treatment of intracranial MPNSTs. Review of the literature and report of a case. RadiatOncol 5: 114.
- Ellis MJ, Cheshier S, Sharma S, Armstrong D, Hawkins C, et al. (2011) Intracerebral malignant peripheral nerve sheath tumor in a child with neurofibromatosis type-1 and middle cerebral artery aneurysm treated with endovascular coil embolization. p. J NeurosurgPediatr 8: 346-352.
- Ouidad Z, Elizabeth F, Laurent Z, Emilie S, Nicolas O, et al. (2013) Chemotherapy for the treatment of malignant peripheral nerve sheath tumors in neurofibromatosis 1: a 10-year institutional review. Orphanet J Rare Dis 8: 127.

